SyntenyLink 🧬
=============
Overview 📖
===========
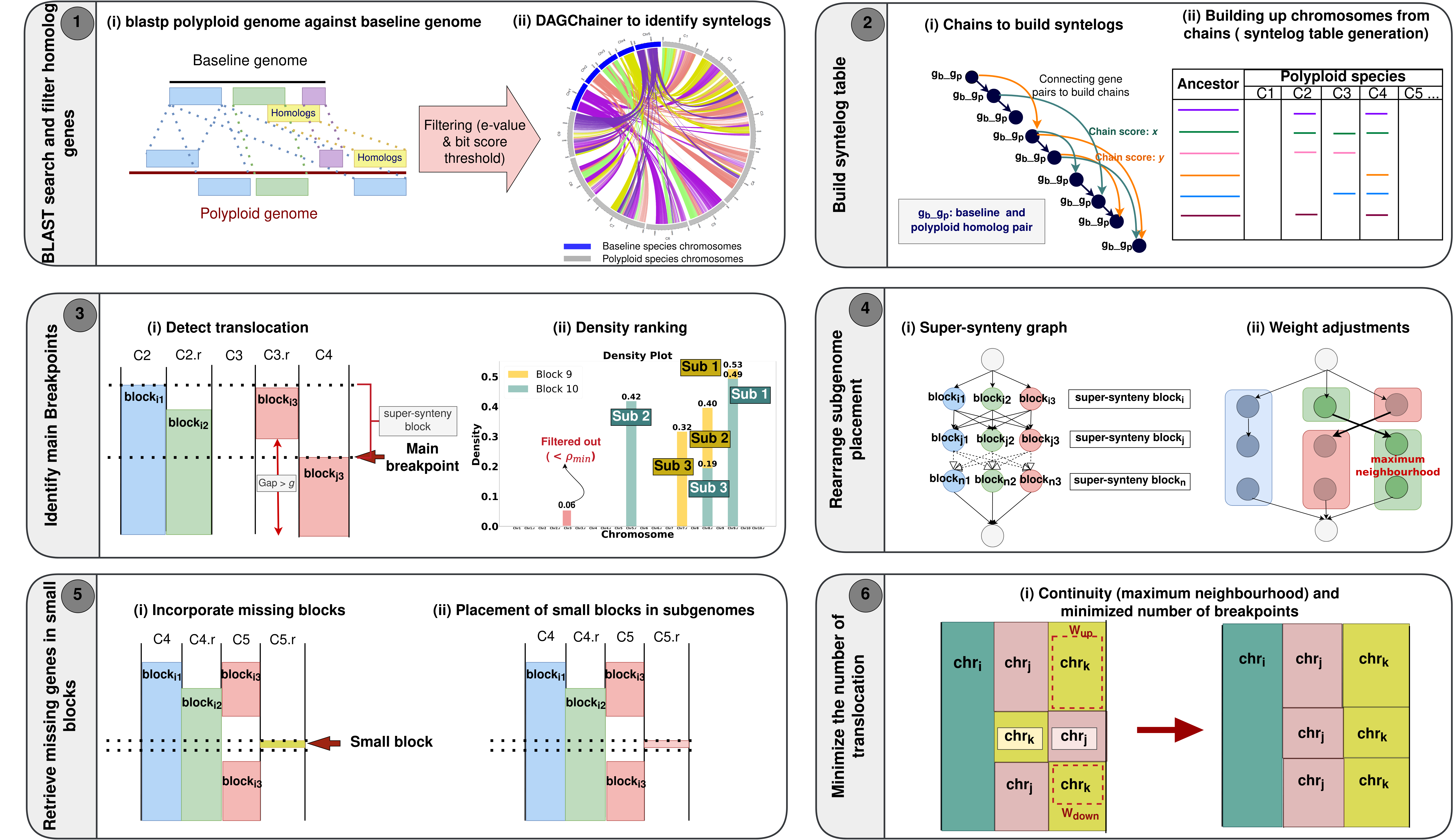
The SyntenyLink package has six major components: the SyntenyLink
algorithm allows users to handle reconstruct subgenomes of polyploid
species more conveniently and to separate the set of genes belong to
each subgenome in the organism with the aid of reference proteomes of
polyploid species and related ancestor. 🌱
All programs are executed using command line options on Linux systems or
Mac OS. Usage or help information are well built into the programs. 💻
All code is copiable, distributable, modifiable, and usable without any
restrictions. Contact:
The following is the list of executable programs
------------------------------------------------
**Main programs (in the Scripts folder)**
- SyntenyLink\_bf.pl
- SyntenyLink\_st.pl
- SyntenyLink\_mbp.py
- SyntenyLink\_wg.py
- SyntenyLink\_sb.py
- SyntenyLink\_mn.py
- main\_script.py
- SyntenyLink\_acc.py
- gap\_threshold\_selection.py
- minimum\_block\_length\_selection.py
Main programs
=============
SyntenyLink\_bf
===============
This program, detects homologs between two species with blastp and
involves a filtering step focusing on bit score and e-value to remove
noise following two criteria: an e-value threshold of less than 1e-20
and a ratio between the bit score and the highest bit score greater than
0.6.
\- Usage Reads in a data file: abc.blast. The abc.blast file holds blast
hits after running blastp between baseline species and polyploid species
of interest:
BraA01g000010.3C AT1G43860.1 74.194 124 14 3 80 186 231 353 9.61e-56 188
Here is a typical parameter setting for generating the abc.blast file:
$ makeblastdb -in ref_pep.fa -dbtype prot -out ref_pep
$ blastall -i query_pep.fasta -p blastp -d ref_pep -m 8 -e 1e-5 -F F -v 5 -b 5 -o abc.blast -a 4
Note: for blastp you need to use protein fasta files of baseline model
and polyploid species of interest. After getting the output blast result
update gene start loci and end loci adding row start values from the bed
file of each species before using SyntenyLink\_bf.py.
It is advised that to make SyntenyLink\_bf generate more reasonable
results, the number of BLASTP hits for a gene should be restricted to
around top 5. When you have abc.blast ready, put them in the same
folder. Then you can simply use:
$ ./SyntenyLink_bf.pl dir/abc.blast
\- Output The execution of SyntenyLink\_bf outputs one blast file
abc\_blast\_filtered.txt, containing filtered blast hits as follows:
BraA01g000010.3C AT1G43860.1 74.194 124 14 3 80 186 231 353 9.61e-56 188
BraA01g000010.3C AT3G04630.1 66.087 115 21 4 194 297 165 272 7.06e-33 125
BraA01g000010.3C AT3G04630.3 66.087 115 21 4 194 297 164 271 7.88e-33 125
SyntenyLink\_st
---------------
This program uses the output after performing synteny analysis using
DAGchainer to build a chain of syntenic genes and compute the score of
each chain. The modified version of filtered results from blastp
(abc\_blast\_filtered\_modified.txt) and DAGchainer
(abc\_synteny.aligncoords) are used to generate a syntelog table in
which the gene chains are incorporated into their corresponding
chromosomes with redundant chains (likely due to gene duplications or
tandem or segmental duplications in the polyploid genome) placed in the
"overlap" column in the syntelog table.
Reads in data file for DAGchainer: abc\_blast\_filtered\_modified.txt.
The abc\_blast\_filtered\_modified.txt file holds the modified
abc\_blast\_filtered.txt file matching the input file format of
DAGchainer:
A1 BraA01g000010.3C 2944 3050 Chr1 AT1G43860.1 16622247 16622597 9.61E-56 188
A1 BraA01g000010.3C 3058 3161 Chr3 AT3G04630.1 1259234 1259503 7.06E-33 125
Note: After getting the abc\_blast\_filtered.txt file update query
start, query end, subject start and subject end values incoorperating
gene locus data from bed files of baseline species and polyploid species
of interest. Then add chromosome which each gene belongs to in the bed
file of each species before using DAGchainer.
Here is a typical parameter setting for generating the
abc\_synteny.aligncoords file:
$ ./run_DAG_chainer.pl -i dir/abc_blast_filtered_modified.txt -s -I
The abc\_synteny.aligncoords file holds pairwise synteny blocks after
running DAGchainer :
## alignment A1 vs. Chr1 Alignment #1 score = 5177.6 (num aligned pairs: 121):
A1 BraA01g026830.3C 17161339 17161504 Chr1 AT1G56580.1 21198405 21198568 2.180000e-109 50
A1 BraA01g026890.3C 17191267 17191318 Chr1 AT1G57550.1 21312544 21312593 3.270000e-24 40
A1 BraA01g026900.3C 17196325 17196618 Chr1 AT1G57610.3 21337612 21337818 8.840000e-106 84
\- Usage Reads in two data files: abc\_blast\_filtered\_modified.txt &
ref\_genelist.txt. The ref\_genelist.txt file holds gff file like
details of baseline species :
AT1G01010 AT1G01010.1 429 Chr1_1 Chr1 1 3631 5899 AT1G01010.1 NAC domain containing protein 1
AT1G01020 AT1G01020.1 245 Chr1_2 Chr1 2 5928 8737 AT1G01020.1 Arv1-like protein
AT1G01030 AT1G01030.1 358 Chr1_4 Chr1 4 11649 13714 AT1G01030.1 AP2/B3-like transcriptional factor family protein
To run SyntenyLink\_st.pl you can simply use:
$ perl SyntenyLink_st.pl -d abc_synteny.aligncoords -g ref_genelist.txt
\- Output The execution of SyntenyLink\_st outputs four output files:
abc\_synteny.success.colinear, abc\_synteny.failed.colinear,
abc\_synteny.chains.passed & abc\_synteny.all.chains.
The abc\_synteny.success.colinear file holds the main output file with
the generated syntelog table in which the gene chains are incorporated
into their corresponding chromosomes with redundant chains (likely due
to gene duplications or tandem or segmental duplications in the
polyploid genome) placed in the "overlap" column in the syntelog table:
BraA01g000010.3C AT1G43860.1 74.194 124 14 3 80 186 231 353 9.61e-56 188
BraA01g000010.3C AT3G04630.1 66.087 115 21 4 194 297 165 272 7.06e-33 125
BraA01g000010.3C AT3G04630.3 66.087 115 21 4 194 297 164 271 7.88e-33 125
The abc\_synteny.failed.colinear file holds the removed chains following
the condition if the number of remaining colinear pairs are less than 6:
A6_Chr1_4 Chr1 AT1G14070 BraA06g010220.3C
A6_Chr1_4 Chr1 AT1G14080 BraA06g010230.3C
A6_Chr1_4 Chr1 AT1G14100 x
A6_Chr1_4 Chr1 AT1G14110 x
A6_Chr1_4 Chr1 AT1G14120 x
A6_Chr1_4 Chr1 AT1G14130 BraA06g010280.3C
A6_Chr1_4 Chr1 AT1G14140 x
A6_Chr1_4 Chr1 AT1G14150 x
A6_Chr1_4 Chr1 AT1G14160 x
A6_Chr1_4 Chr1 AT1G14170 x
A6_Chr1_4 Chr1 AT1G14180 x
A6_Chr1_4 Chr1 AT1G14185 x
The abc\_synteny.chains.passed file holds the ids of set of passed
chains with collinear pairs greater than 6:
A7_Chr1_1
A8.r_Chr1_1
A9.r_Chr1_1
A2_Chr1_1
A6_Chr1_1
A7.r_Chr1_1
The abc\_synteny.all.chains file holds the all chains identified from
DAGchainer:
A1_Chr1_1 A1 BraA01g026830.3C 17161339 17161504 Chr1 AT1G56580.1 21198405 21198568 2.180000e-109 50
A1_Chr1_1 A1 BraA01g026890.3C 17191267 17191318 Chr1 AT1G57550.1 21312544 21312593 3.270000e-24 40
A1_Chr1_1 A1 BraA01g026900.3C 17196325 17196618 Chr1 AT1G57610.3 21337612 21337818 8.840000e-106 84
SyntenyLink\_mbp
----------------
This program identifies the main breakpoints of translocations in the
abc\_synteny.success.colinear file. It is is designed to detect
fractionation gaps larger than a gap threshold. Synteny blocks capped by
two breakpoints are grouped as a "super-synteny block" where gaps
smaller than the gap threshold could exist within the super block. The
input to this algorithm includes two parameters gap threshold, a minimum
block length, and a data file syntelog table generated from Step 2,
where collinear blocks are placed into the chromosomes of the organism.
Next, the gene density of super-synteny blocks in each chromosome is
calculated. This algorithm disregards densities below threshold density
value. We selected a threshold density value of 0.1 in here. The
chromosomes with gene density greater than 0.1 are ranked by the density
of the blocks within the chromosome and assigned into candidate 𝑚
subgenomes, where 𝑚 denotes the ploidy level or the number of subgenomes
to be reconstructed.
\- Usage No input parameters or read in data files.
Note: You need to select the most suitable gap threshold and minimum
block length for your data by running the gap\_threshold\_selection.py
and minimum\_block\_length\_selection.py scripts.
Here is a typical parameter setting for obtaining optimal gap threshold
and block length values for you data:
$ python3 gap_threshold_selection.py -i abc_synteny.success.colinear
Then get the optimal gap threshold value from the console and run the
following command:
$ python3 minimum_block_length_selection.py -i abc_synteny.success.colinear -g <output gap threshold value>
\- Output Optimal gap threshold and block length values are printed to
the console.
\- Output The execution of SyntenyLink\_mbp outputs two output files:
Super\_synteny\_block\_output.xlsx and
Super\_synteny\_bl\_sub\_placement\_density.xlsx.
The Super\_synteny\_block\_output.xlsx file holds the generated
super-synteny blocks before removing the noise taking density threshold
into account:
Block no. Block_start Block_end Row start # Row end # # genes in block N1 N1.r N2 N2.r N3 N3.r N4 N4.r N5 N5.r N6 N6.r N7 N7.r N8 N8.r
1 AT1G01010 AT1G01560 0 57 58 0.0 0.0 0.0 0.3684210526315789 0.0 0.6666666666666666 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0
2 AT1G01570 AT1G02205 58 123 66 0.0 0.0 0.43283582089552236 0.0 0.0 0.4626865671641791 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0
3 AT1G02210 AT1G14900 124 1491 1368 0.005113221329437546 0.0 0.3951789627465303 0.005843681519357195 0.3915266617969321 0.0 0.0577063550036523 0.1891891891891892 0.0 0.0 0.07596785975164354 0.26004382761139516 0.0 0.0 0.0 0.0
The Super\_synteny\_bl\_sub\_placement\_density.xlsx file holds the
subgenome placements of super-synteny blocks based on gene density,
after removing the noise taking density threshold into account:
Block no. Block_start Block_end Row start # Row end # # genes in block N1 N1.r N2 N2.r N3 N3.r N4 N4.r N5 N5.r N6 N6.r N7 N7.r N8 N8.r N9 N9.r Non_zero subgenome1 subgenome2 subgenome3
1 AT1G01010 AT1G11860 0 1161 1162 0 0 0 0 0 0 0 0 0.544358312 0 0 0 0 0 0.354005168 0.308354866 0 0 3 N5 N8 N8.r
2 AT1G11870 AT1G13420 1162 1328 167 0 0 0 0 0 0 0 0 0 0.523809524 0 0 0 0 0.386904762 0.43452381 0 0 3 N5.r N8.r N8
3 AT1G13430 AT1G19470 1329 1957 629 0 0 0 0 0 0 0 0 0.598412698 0 0 0 0 0 0.33968254 0.422222222 0 0 3 N5 N8.r N8
SyntenyLink\_wg
---------------
This program uses a weighted direct graph to dynamically link blocks
produced from Step 3 that are most likely to be in a subgenome using a
combined information of fractionation and substitution patterns as well
as continuity of gene chains. This algorithm takes two input parameters
a1 and a2.
\- Usage Reads in two data files: abc\_synteny.all.chains and
abc\_blastn.blast. Two input parameters:a1 and a2.
Note: You need to generate abc\_blastn.blast file by running nucleotide
blast using cds fasta files of baseline species and polyploid species of
interest.
The abc\_blastn.blast file holds blast hits after running blastn between
baseline species and polyploid species of interest:
BraA01g000010.3C AT1G43860.1 78.706 371 25 6 239 558 692 1059 7.94e-91 335
Here is a typical parameter setting for generating the abc\_blastn.blast
file:
$ makeblastdb -in ref_cds.fa -dbtype prot -out ref_cds
$ blastall -i query_cds.fasta -p blastn -d ref_cds -m 8 -e 1e-5 -F F -v 5 -b 5 -o abc_blastn.blast -a 4
Note: You need to select the most suitable a1 and a2 parameter values
for your data, which accounts for adjusting weights of the graph.
\- Output The execution of SyntenyLink\_wg outputs number of output
files matching the number of subgenomes in the species of interest:
Super\_synteny\_graph\_nodes\_sub{k}.xlsx. There will be m number of
files. k represents the corresponding subgenome number.
The Super\_synteny\_graph\_nodes\_sub{k}.xlsx file holds the nodes
belong to each subgenome after traversing the graph:
Row start # Row end # subgenome1 subgenome2 subgenome3
0 123 N10.r N9 N8.r
124 698 N10 N8.r N9.r
699 1029 N6 N9.r N8.r
SyntenyLink\_sb
---------------
This program retrieves genes "hidden" in small blocks that are missed in
previous steps. Specically, we consider the chromosome blocks with
densities lower than d\<8= that are removed in previous steps in the
generation of small blocks because these blocks may be more likely to
exhibit ipping of synteny blocks between the forward and reverse
strands resulting from segmental duplication. Following the generation
of small blocks, small synteny blocks are incorporated into the
corresponding super-synteny blocks, taking into account the subgenome
placement of the super-synteny blocks as a reference.
\- Usage No read in files or parameters.
\- Output The execution of SyntenyLink\_sb outputs number of output
files: modified\_chr\_names{k+1}.xlsx, subgenome\_placement\_blocks.all{k+1}.xlsx, subgenome\_placement\_blocks.all.xlsx. k represents the corresponding subgenome number.
The modified\_chr\_names{k+1}.xlsx file holds the
replaced gene id\'s with there corresponding chromosome names which they
bellong to if they are not already placed inside the subgenome columns (to detect the genes that has been removed based on density threshold):
Gene_id N1 N1.r N2 N2.r N3 N3.r N4 N4.r N5 N5.r N6 N6.r N7 N7.r
AT1G01900 0 0 0 0 0 0 0 0 0 1 0 0 0 0
AT1G01910 0 0 0 0 0 0 0 0 0 0 0 1 0 0
AT1G01920 0 0 0 0 0 0 0 0 0 0 0 1 0 0
AT1G01930 0 0 0 0 0 0 0 0 0 0 0 1 0 0
AT1G01940 0 0 0 0 0 0 0 0 0 0 0 1 0 0
AT1G01950 0 0 0 0 0 0 0 0 0 0 N6 0 0 0
AT1G01960 0 0 0 0 0 0 0 0 0 0 0 1 0 0
AT1G01970 0 0 0 0 0 0 0 0 0 1 0 0 0 0
The subgenome\_placement\_blocks.all{k+1}.xlsx file holds the placement
of genes in subgenomes optimal for each subgenome considering the subgenome separation optimal for each subgenome in step 4:
Row start # Row end # subgenome1 subgenome2 subgenome3
0 123 N10.r N9 N8.r
124 698 N10 N8.r N9.r
699 1029 N6 N9.r N8.r
The subgenome\_placement\_blocks.all.xlsx file holds the final placement
of genes in subgenomes in step 5:
Row start # Row end # subgenome1 subgenome2 subgenome3
0 123 N10.r N9 N8.r
124 698 N10 N8.r N9.r
699 1029 N6 N9.r N8.r
SyntenyLink\_mn
---------------
Aimed to optimize the subgenome placements for each block (a row in the
subgenome matrix) based on neighbourhood considerations in a subgenome,
maximize the number of continuous blocks/rows from the same chromosome.
The algorithm utilizes two window sizes, up and down, which determine
the number of blocks above and below a given block, to define a
neighbourhood.
\- Usage No read in files Two input parameters: wup wdwn.
Note: You need to select the most suitable window up and window down
parameters.
\- Output The execution of SyntenyLink\_sb outputs number of output
files: Final\_subgenome\_placement\_result.xlsx, Final\_result.xlsx.
The Final\_subgenome\_placement\_result.xlsx file holds the final
placement of chromosome blocks in subgenomes:
Row start # Row end # subgenome1 subgenome2 subgenome3
0 75 N10.r N9 N8.r
76 78 N10.r N9.r N8.r
79 123 N10.r N9 N8.r
124 698 N10 N9.r N8.r
699 1035 N6 N9.r N8.r
1036 1036 N6 N9 N8.r
The Final\_result.xlsx file holds the final placement of genes inside
blocks in subgenomes:
subgenome1 subgenome2 subgenome3
BraA10g000880.3C x x
BraA10g000870.3C x x
BraA10g000860.3C x x
BraA10g000850.3C x x
BraA10g000830.3C BraA09g065940.3C x
BraA10g000820.3C x x
x BraA09g065960.3C x
BraA10g000790.3C x x
BraA10g000780.3C BraA09g065970.3C x
BraA10g000770.3C BraA09g065980.3C x
main\_script
------------
This holds the main script that runs all the above scripts in order. It
takes in the following parameters: -i input\_file -g gap\_threshold -m
minimum\_block\_length -n number\_of\_subgenomes -gt ground\_truth\_file
-c chains\_file -bl blastn\_file -a1 adjusment1 -a2 adjusment2 -ws1
window\_size\_subgenome1 -ws2 window\_size\_subgenome2 -ws3
window\_size\_subgenome3 -wup window\_up -wdwn window\_down
Note: Before running main\_script.py, you need to run the
SyntenyLink\_bf.pl and SyntenyLink\_st.pl scripts
To run SyntenyLink\_sb.py you can simply use:
$ python3 main_script.py -i abc_synteny.success.colinear -g -m -n -gt abc_groundtruth.xlsx -c abc_synteny.all.chains -bl abc_blastn.blast -a1 -a1 -a1 -a2 -a2 -a2 -ws1 -ws2 -ws3 -wup -wdwn
\- Output The execution of main\_script outputs all the output files of
the above scripts in the same directory as the input file.
SyntenyLink\_acc
----------------
This holds the script that calculates the accuracy of the final
placement of genes in subgenomes when there is a ground truth file.
\- Usage One read in file: abc\_groundtruth.xlsx
To run SyntenyLink\_sb.py you can simply use:
$ python3 SyntenyLink_acc.py abc_groundtruth.xlsx
\- Output The execution of SyntenyLink\_sb prints the subgenome
placemnet accuracy of each subgenome:
Exact match percentage for subgenome1: 84.15%
Exact match number for subgenome1: 11490
Missing genes for subgenome1: 2164
Exact match percentage for subgenome2: 61.61%
Exact match number for subgenome2: 6062
Missing genes for subgenome2: 3778
Exact match percentage for subgenome3: 65.30%
Exact match number for subgenome3: 5635
Missing genes for subgenome3: 2995
License 📜
===============
This project is licensed under the [MIT License](LICENSE)